Researchers of the Institute for Software Technology, together with colleagues from the Karlsruhe Institute of Technology and the Forschungszentrum Jülich found a computational method to predict RNA structures with the help of deepl-learning-methods.
The interdisciplinary research team developed a computational method to predict RNA structures as part of the project ProFiLe. A task with potentially far-reaching impact, since RNA is present in all living organisms, and it plays a crucial role in various biological processes, including protein synthesis, gene regulation, and more. The results are impressive as the conceptional advance could prove a breakthrough in decreasing the sequence-structure gap for RNA and the underlying method is generalizable to other tasks.
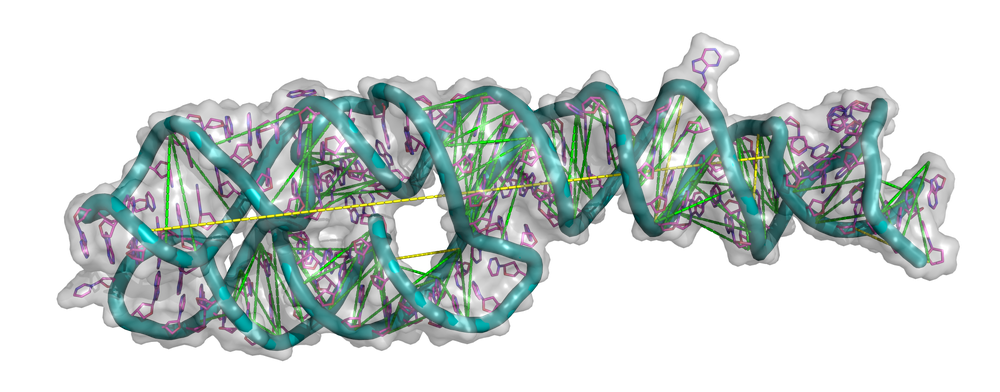
Visualization of an RNA molecule (PDB: 3ndb)
Credit:
DLR et al. CC-BY https://www.nature.com/articles/s42003-023-05244-9.pdf
Comparison of the contact map with a restrained backbone and a free backbone.
Predicting RNA folding is a complex problem, as the molecules can fold into a large number of possible three-dimensional structures. Experimentally extracting information out of RNA is biochemically difficult, and this leads to the data base for starting the deep learning cycle being somewhat narrow. Utilizing the limited data available, the research group focused on predicting spatial adjacencies, known as contact maps, as a proxy for 3D structure.
The study delves into the realm of self-supervised learning for RNA multiple sequence alignments, with a specific focus on predicting contacts from latent attention maps. The introduction of boosted decision trees significantly enhanced the quality of contact predictions, which were further refined through fine-tuning of the pretrained backbone.